
Conformational Searching with CONFLEX
In Example 4.8 of Exploring Chemistry with Electronic Structure Methods (beginning on p.164), we predict the NMR chemical shifts with respect to TMS for the carbon atoms in 2,2,4-trimethyl-1,3-pentanediol. This molecule has many conformations, and it is necessary to take that into consideration when predicting its NMR spectrum. Accordingly, this study proceeds in the following way:
- We begin with a conformational search to locate all of the structures of potential interest.
- We optimize each conformation with the B3LYP/3-21G model chemistry, following the procedure in the example in the book (the CONFLEX study described here used a different basis set for this step).
- We examine the resulting optimized structures and eliminate duplicates. We then perform Opt+Freq calculations for each remaining structure using the B3LYP/6-311+G(2d,p) model chemistry.
- We once again eliminate duplicate structures as well as those which contribute less than about 0.5% to the Boltzmann distribution. We run NMR calculations on the remaining structures after verifying that they are minima.
- Finally, we predict the chemical shift for each carbon atom by Boltzmann-averaging the results of the NMR jobs.
![]()
The CONFLEX conformational analysis system can be used to perform the conformational search for this study (see conflex.net), as well as for other research requiring computational searching. In this article, we demonstrate the use of this software on this problem.
CONFLEX can open Gaussian input files directly, so we open the e4_08_start.gjf file (right click here to download). The CONFLEX display will appear like this:
The molecular structure appears in the large window on the right. We now save the file as an MDL file prior to running the conformational search:
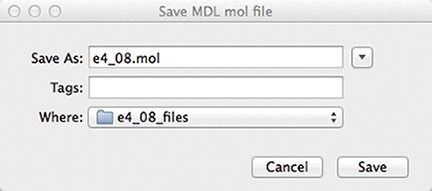
We now start the conformational search by selecting the CONFLEX item from the Calculation menu. The following dialog appears at the top of the molecule display:
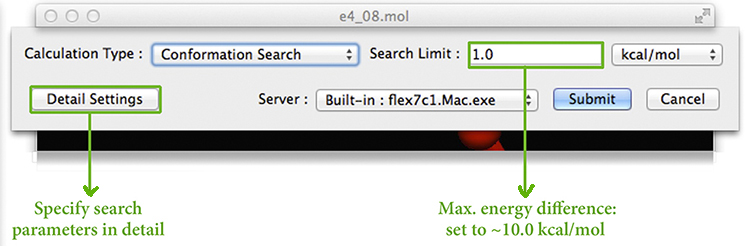
We specify that we want to perform a Conformational Search in the Calculation Type menu. The Search Limit field specifies the maximum energy difference between the lowest energy and highest energy conformations. It allows you to control the completeness of the search as well as its duration: the more completely the conformational space is searched, the longer the computation takes.
The best value to specify for this parameter depends on your purpose and on the size of the molecule:
- If your goal is to locate the most stable conformation, then a value of 3-5 kcal/mol will be sufficient for a molecule of this size we are considering.
- If you want find all conformations, then you will need a larger value. For a molecule of this size, a value of 13-15 kcal/mol should be sufficient. Be aware, however, that this range may be impractical for very large molecules.
The Detail Settings button can be used to specify additional aspects of the conformation search. However, for our case, the defaults work fine, so we can submit the job at this point. We chose to run the job on the local computer, but we could specify a remote host via the Server popup menu.
The CONFLEX Job Manager will now open:
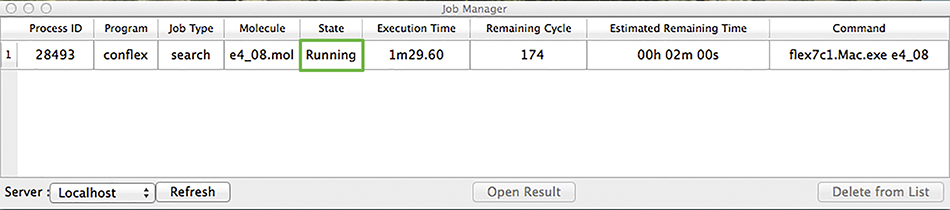
The various columns of this table give information about each running calculation. In this example, our conformational search is the only one listed. It is currently running, as indicated in the State column. When the job is complete, the state will be listed as Finished. At this point, we can double click on the job to open its results (or select the job and click Open Result). The following display will result:
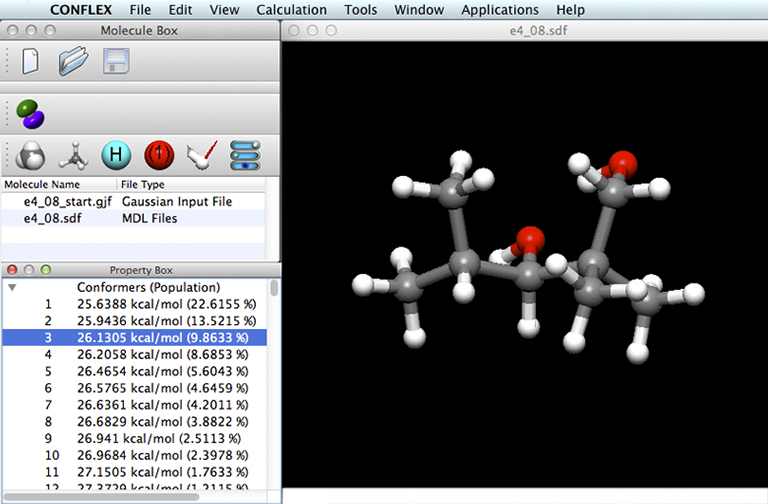
A new molecule display has been opened for the .SDF file: the results file type for a conformational search. In addition, the Property Box panel has been opened. It contains the list of conformations that were located by the search. Each line contains the conformation number, its predicted energy, folowed by that conformation’s contribution to the Boltzmann average in parentheses. The molecule display shows the structure of the selected conformation (number 3 in the preceding example).
The CONFLEX software performs conformational searches using strategies designed to exhaustively search the conformational space of the target molecule, using a combination of stepwise perturbations and perturbation modes which mimic thermal vibrations:
You can examine the Conformers List to determine how extensively the space was searched given the specified energy range and other parameters. Use the Conformers List item on the Tools menu to access it:
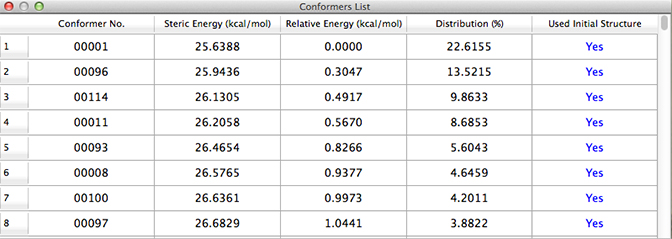
The table lists the conformations in order to increasing energy, giving the conformation number, predicted energy, energy difference from the lowest energy conformation and contribution to the Boltzmann distribution. The final column, Used Initial Structure, indicates whether the structure was used as a starting point for additional iterations during the course of the search. When the value is Yes for all conformations, then a full iterative procedure has been completed. The CONFLEX documentation explains it this way:
“Yes” indicates that a conformer was selected as starting structure during conformation search. If all conformers within Search Limit have a “Yes” flag, CONFLEX Conformation Search has finished.
Here is the final portion of the conformation list from our search:
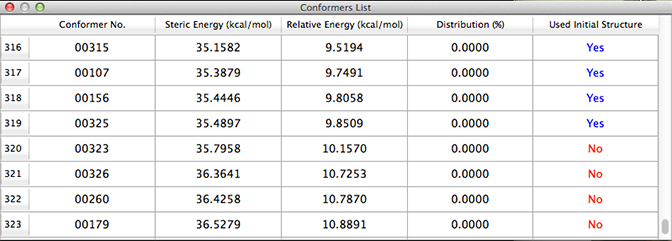
The last few conformations have a value of No in the final column, so technically this search is not completely exhaustive. However, the relative energies of the highest energy conformations–more than 10 kcal/mol–indicate that they will contribute negligibly to the NMR results, so we can consider this conformational search to be sufficient.
The next step is to optimize each conformation with Gaussian using the B3LYP/3-21G model chemistry. If a remote server has been specified to the CONFLEX software, then we can do so using a single step. We begin by selecting all of the conformations in the Property Box. Next, we select GAUSSIAN from the Calculation menu, which results in the following dialog appearing at the top of the molecule window:
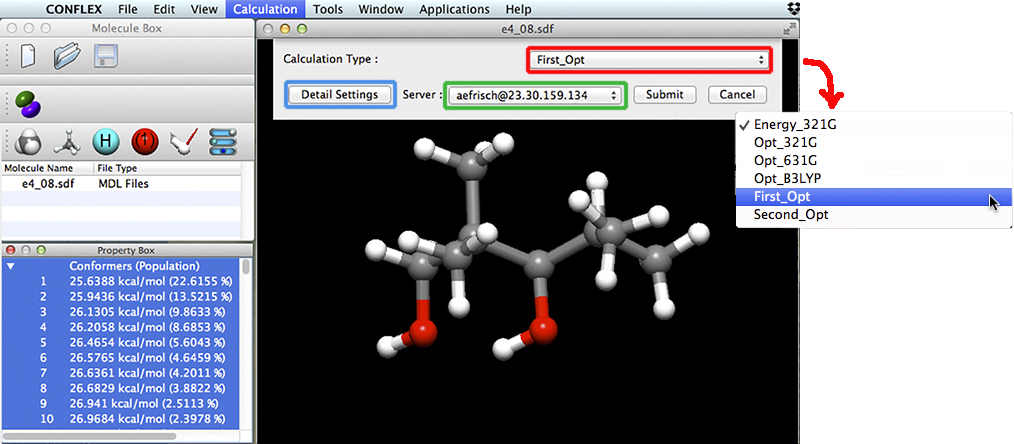
We specify the remote server by selecting it from the Server popup menu (indicated in green above). We can use the Detail Settings button to access the various settings for the Gaussian calculations. However, we have previously defined and saved a calculation scheme with the parameters we want, giving it the name First_Opt. We select it from the Calculation Type menu, and the saved specifications are automatically used for each optimization calculation.
Once we click Submit, a batch job containing the desired set of calculations is sent to the queueing system on the remote server. The resulting job is listed in the Job Manager. The following example shows how such a job would look for one batch queueing system and a customized Job Manager display:
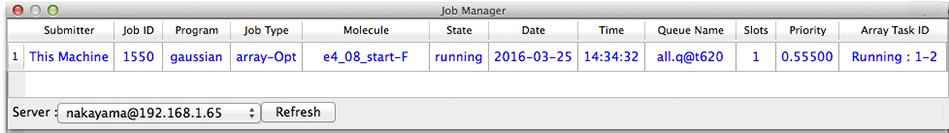
Once the display indicates that the job is complete, double clicking on it will cause the result files to be acquired from the remote server. The total energy for each optimized structure will also be extracted, and the set of energies will be listed in the Property Box. Be aware that these values will be listed in the same order as for the initial conformational search; they will probably not be ordered by increasing energy.
We will go on to run a second set of Opt+Freq calculations on the unique structures within the resulting set, using the B3LYP/6-311+G(2d,p) model chemistry. We will use a previously set up calculation scheme named Second_Opt. Once these jobs are complete, we will run NMR calculations on the energetically significant conformations. Finally, we will Boltzmann average the predicted NMR chemical shifts via an external spreadsheet.
This note was prepared by Dr. Naofumi Nakayama (CONFLEX Corp.) and Dr. Æleen Frisch (Exponential Consulting).

Related Posts
Tags
Share This





